
Population Structure and Diversity in European Honey Bees (Apis mellifera L.)—An Empirical Comparison of Pool and Individual Whole-Genome Sequencing

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
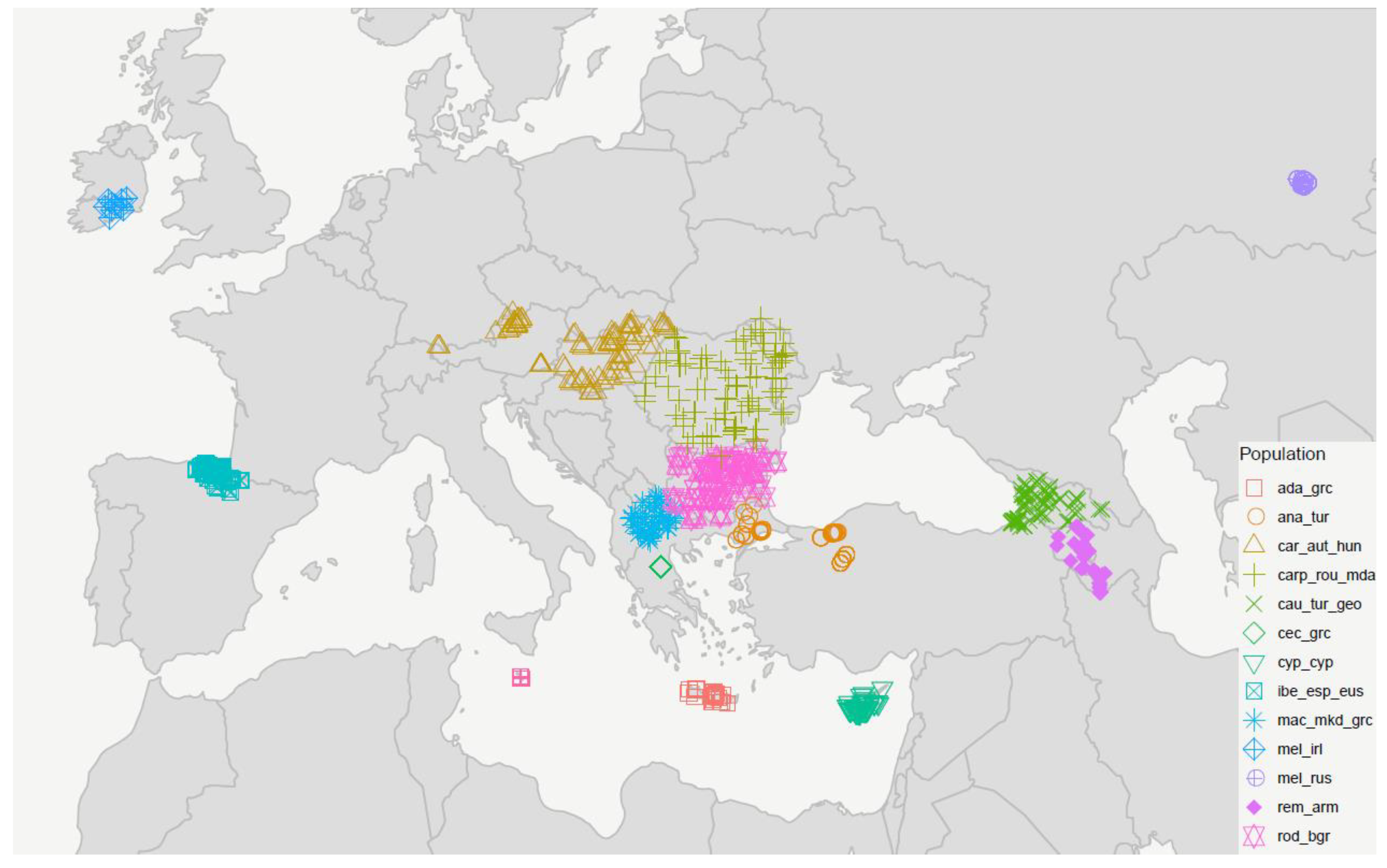
2.1. Sampling
2.2. DNA Extraction and Sequencing
2.3. Sequencing Data Processing
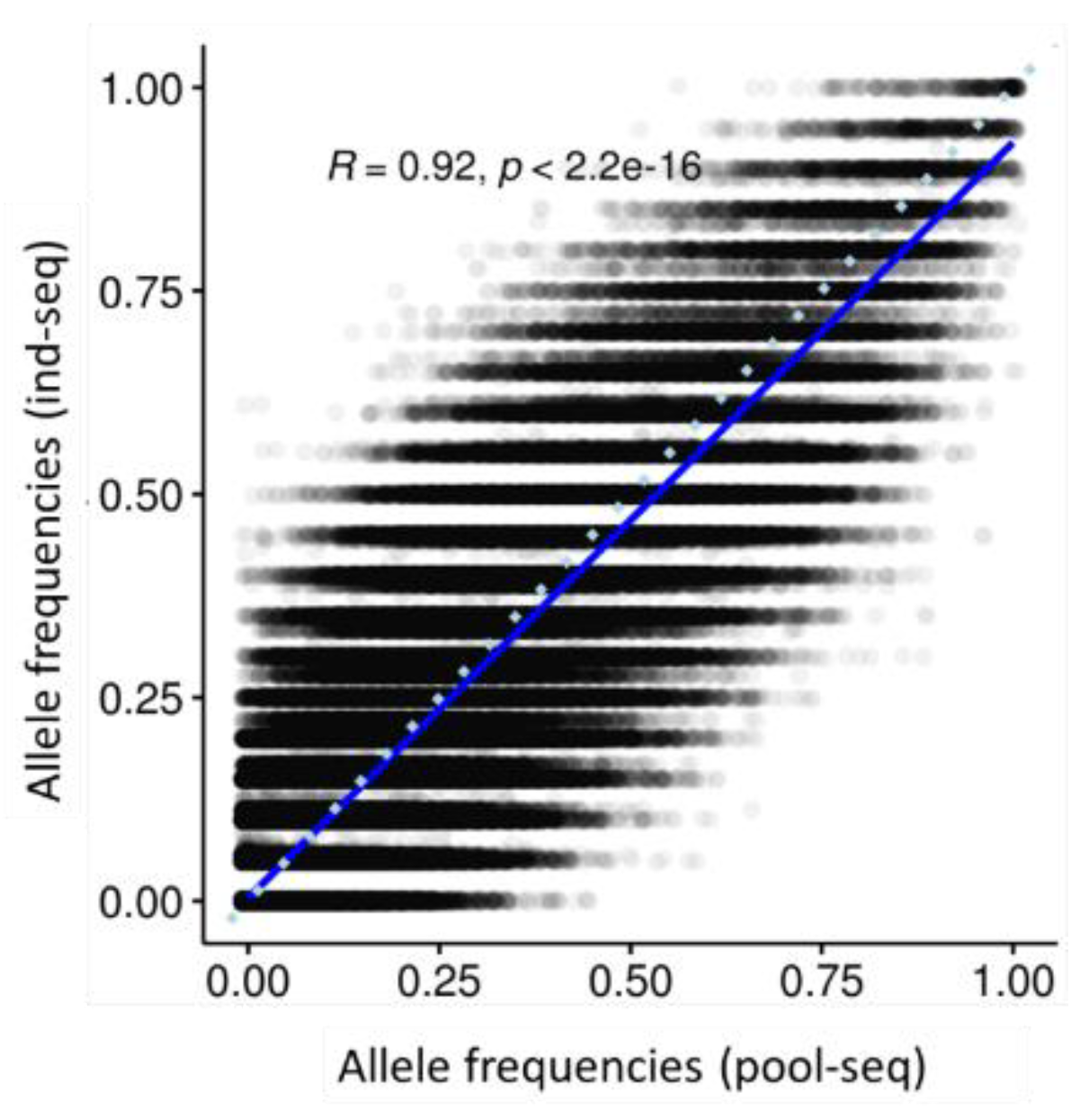
2.4. Allele Frequency Correlation
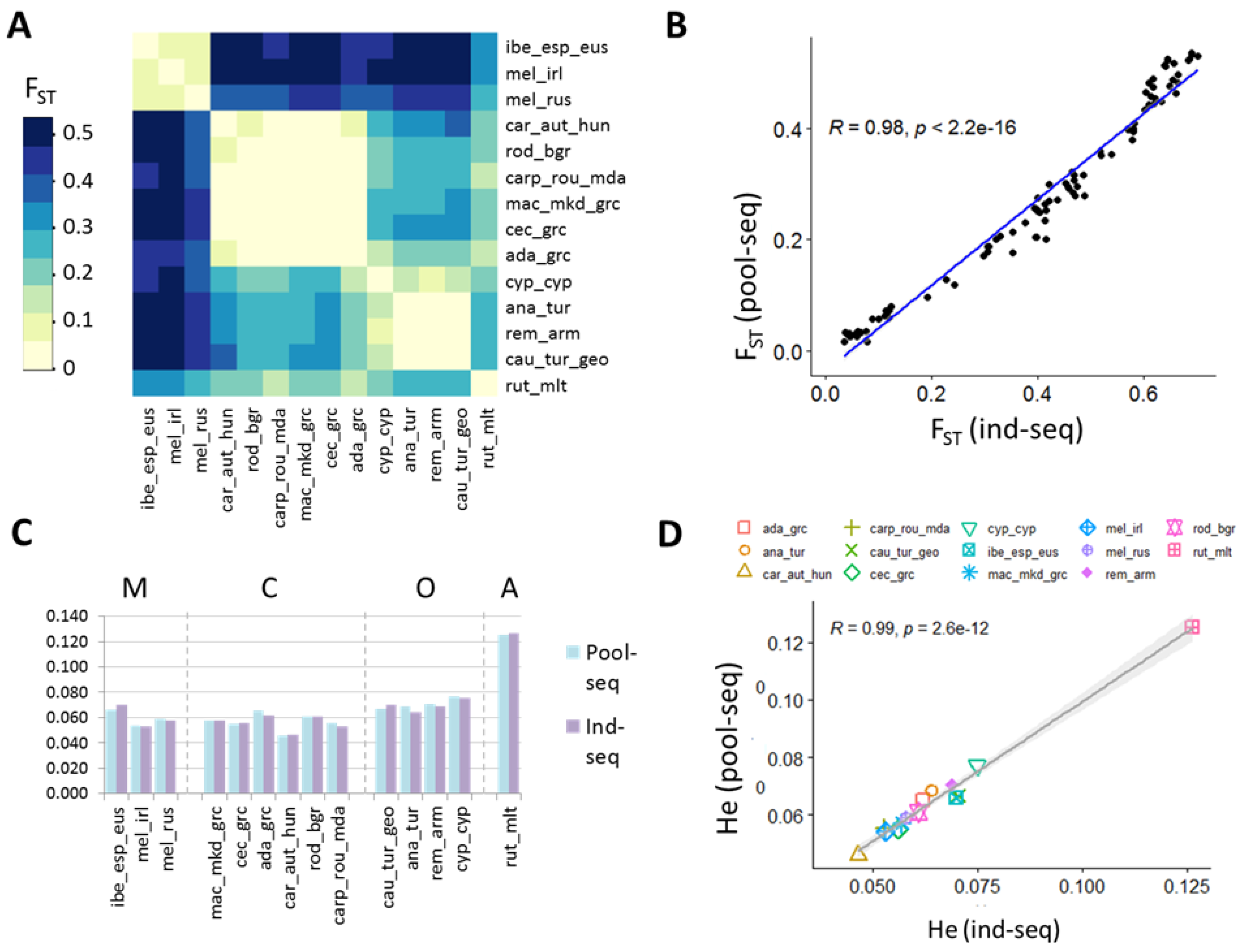
2.5. Genetic Diversity between and within Populations
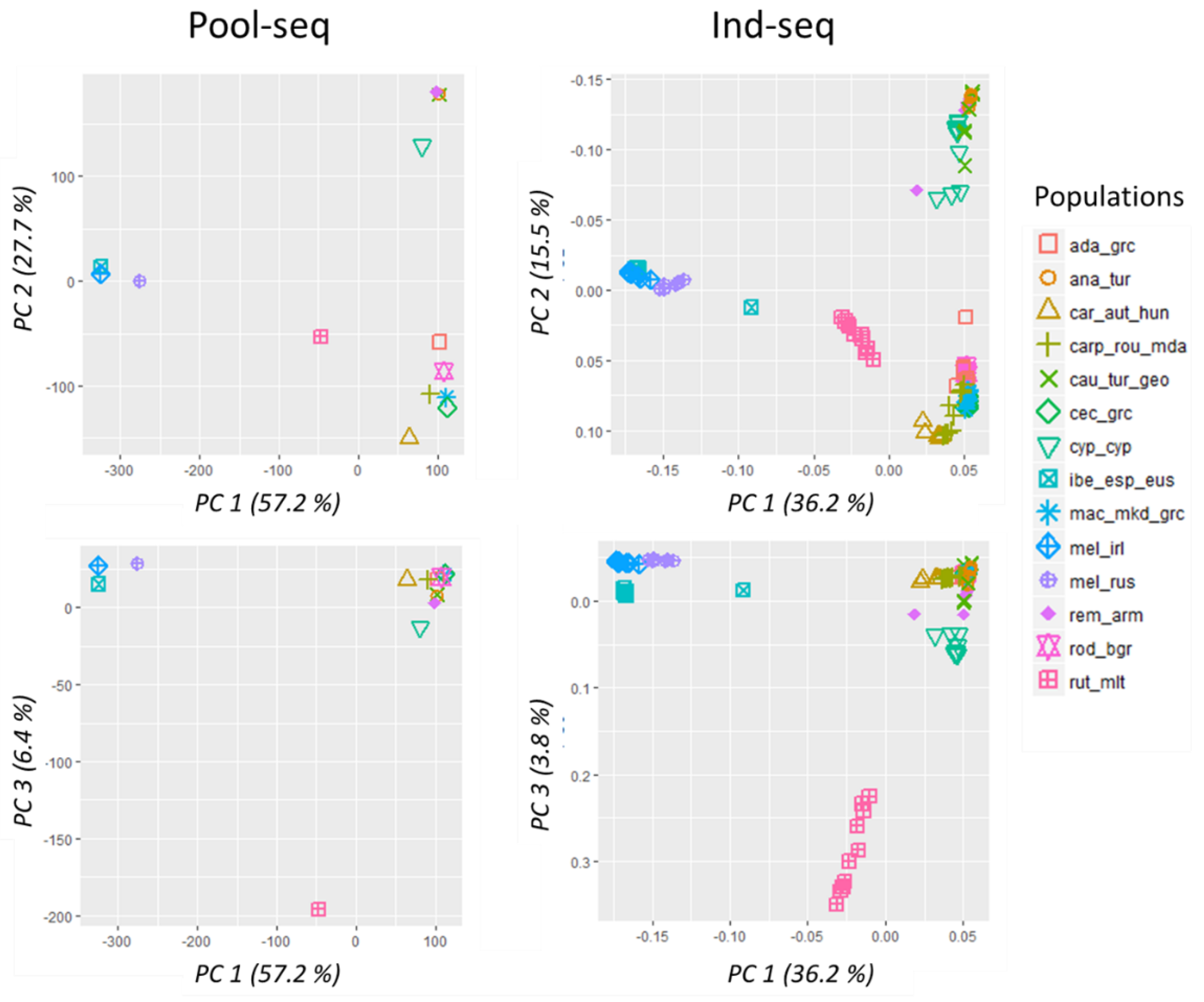
2.6. Population Structure
3. Results
3.1. Sequence Data and Variants
3.2. Allele Frequency Correlation
3.3. Genetic Diversity between and within Populations
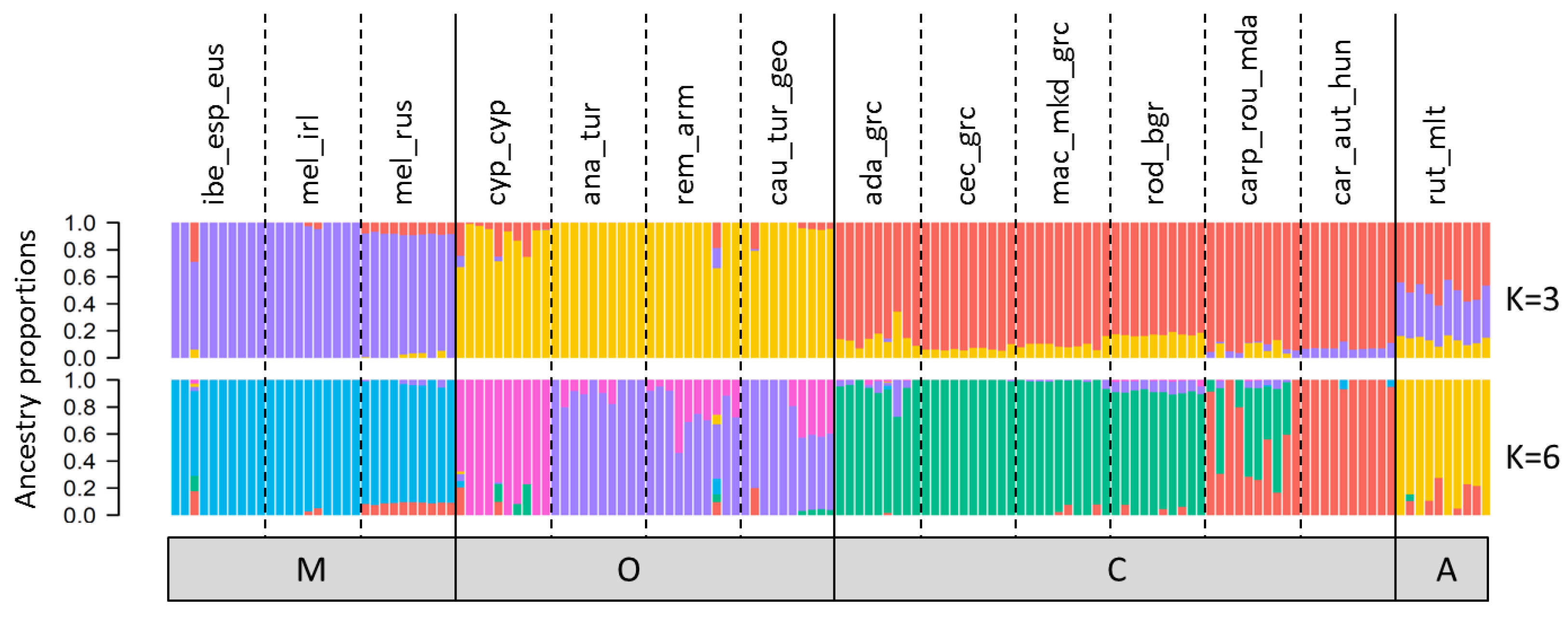
3.4. Population Structure
4. Discussion
4.1. The Limited Ability of Pool-Seq to Identify Low-Frequency Variants
4.2. Sampling and the Importance of Previous Knowledge for Pool-Seq
4.3. Near Identical Inference of Population Structure and Diversity by Pool-Seq and Ind-Seq
4.4. Both Approaches Compare Well to Established Studies
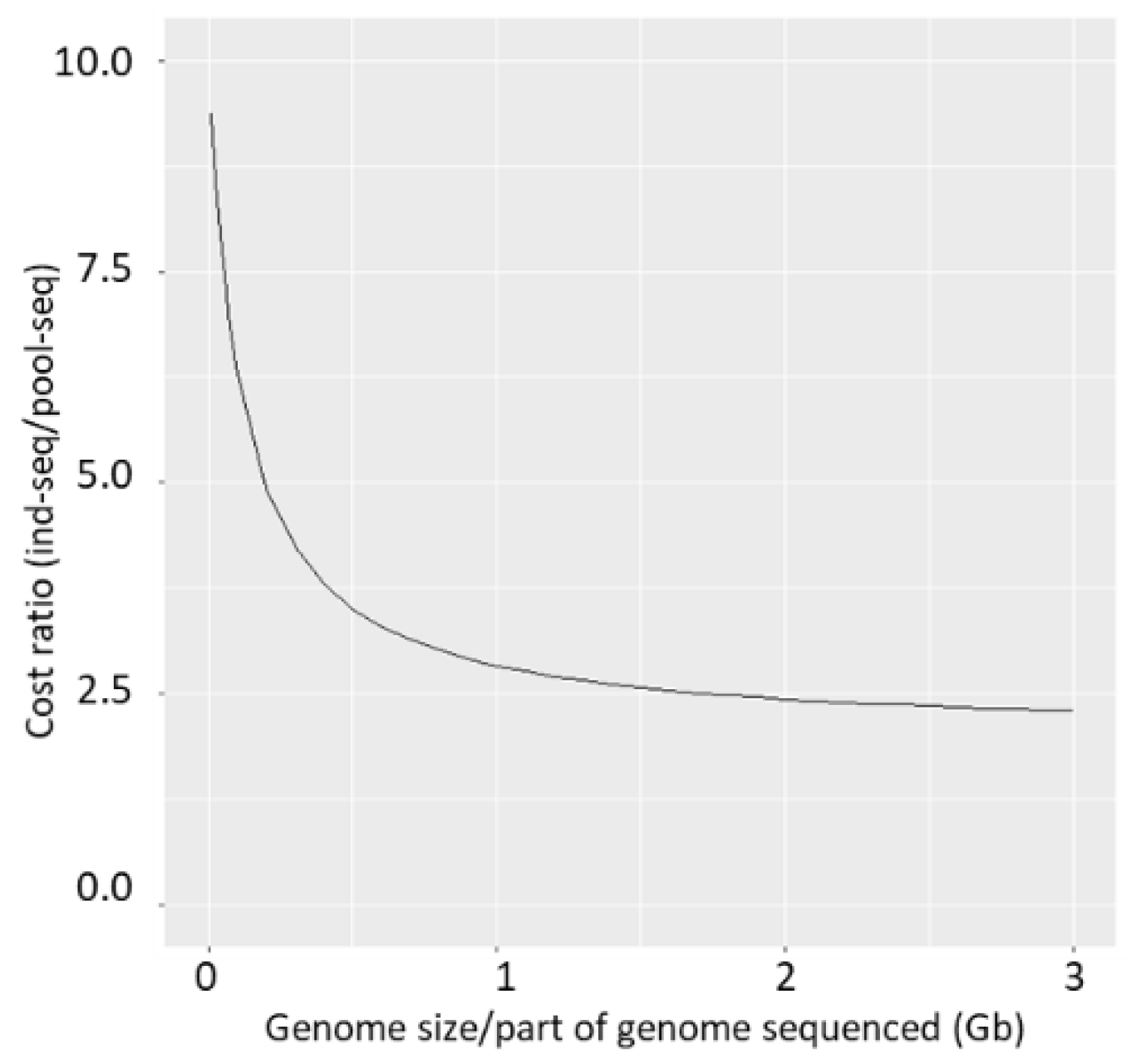
4.5. The Cost-Benefit Ratio between Pool-Seq and Ind-Seq
4.6. Additional Insights from Ind-Seq
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Allendorf, F.W.; Luikart, G.; Aitken, S.N. Conservation and the Genetics of Populations, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Luikart, G.; Kardos, M.; Hand, B.K.; Rajora, O.; Aitken, S.; Hohenlohe, P.A. Population Genomics: Advancing Understanding of Nature; Springer: Cham, Germany, 2018. [Google Scholar]
- Foote, A.D.; Vijay, N.; Avila-Arcos, M.C.; Baird, R.W.; Durban, J.W.; Fumagalli, M.; Gibbs, R.A.; Hanson, M.B.; Korneliussen, T.S.; Martin, M.D.; et al. Genome-culture coevolution promotes rapid divergence of killer whale ecotypes. Nat. Commun. 2016, 7, 11693. [Google Scholar] [CrossRef] [Green Version]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloglu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef] [Green Version]
- Dorant, Y.; Benestan, L.; Rougemont, Q.; Normandeau, E.; Boyle, B.; Rochette, R.; Bernatchez, L. Comparing Pool-seq, Rapture, and GBS genotyping for inferring weak population structure: The American lobster (Homarus americanus) as a case study. Ecol. Evol. 2019, 9, 6606–6623. [Google Scholar] [CrossRef] [Green Version]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Futschik, A.; Schloetterer, C. The Next Generation of Molecular Markers from Massively Parallel Sequencing of Pooled DNA Samples. Genetics 2010, 186, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Schloetterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals-mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef]
- Cutler, D.; Jensen, J. To Pool, or Not to Pool? Genetics 2010, 186, 41–43. [Google Scholar] [CrossRef] [Green Version]
- Gautier, M.; Foucaud, J.; Gharbi, K.; Cezard, T.; Galan, M.; Loiseau, A.; Thomson, M.; Pudlo, P.; Kerdelhue, C.; Estoup, A. Estimation of population allele frequencies from next-generation sequencing data: Pool-versus individual-based genotyping. Mol. Ecol. 2013, 22, 3766–3779. [Google Scholar] [CrossRef]
- Saelao, P.; Simone-Finstrom, M.; Avalos, A.; Bilodeau, L.; Danka, R.; de Guzman, L.; Rinkevich, F.; Tokarz, P. Genome-wide patterns of differentiation within and among US commercial honey bee stocks. BMC Genom. 2020, 21, 704. [Google Scholar] [CrossRef]
- Yunusbaev, U.B.; Kaskinova, M.D.; Ilyasov, R.A.; Gaifullina, L.R.; Saltykova, E.S.; Nikolenko, A.G. The Role of Whole-genome Research in the Study of Honey Bee Biology. Russ. J. Genet. 2019, 55, 778–787. [Google Scholar] [CrossRef]
- Rellstab, C.; Zoller, S.; Tedder, A.; Gugerli, F.; Fischer, M.C. Validation of SNP Allele Frequencies Determined by Pooled Next-Generation Sequencing in Natural Populations of a Non-Model Plant Species. PLoS ONE 2013, 8, e80422. [Google Scholar] [CrossRef]
- Wallberg, A.; Han, F.; Wellhagen, G.; Dahle, B.; Kawata, M.; Haddad, N.; Paulino Simoes, Z.L.; Allsopp, M.H.; Kandemir, I.; De la Rua, P.; et al. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat. Genet. 2014, 46, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Momeni, J.; Parejo, M.; Nielsen, R.O.; Langa, J.; Montes, I.; Papoutsis, L.; Farajzadeh, L.; Bendixen, C.; Cauia, E.; Charriere, J.-D.; et al. Authoritative subspecies diagnosis tool for European honey bees based on ancestry informative SNPs. BMC Genom. 2021, 22, 101. [Google Scholar] [CrossRef]
- Ruttner, F. Biogeography and Taxonomy of Honeybees; Springer: Berlin/Heidelberg, Germany, 1988; pp. 163–257. [Google Scholar]
- Garnery, L.; Cornuet, J.M.; Solignac, M. Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol. Ecol. 1992, 1, 145–154. [Google Scholar] [CrossRef]
- Sheppard, W.S.; Arias, M.C.; Grech, A.; Meixner, M.D. Apis mellifera ruttneri, a new honey bee subspecies from Malta. Apidologie 1997, 28, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Kandemir, I.; Kence, M.; Sheppard, W.S.; Kence, A. Mitochondrial DNA variation in honey bee (Apis mellifera L.) populations from Turkey. J. Apic. Res. 2006, 45, 33–38. [Google Scholar] [CrossRef]
- De la Rua, P.; Jaffe, R.; Dall’Olio, R.; Munoz, I.; Serrano, J. Biodiversity, conservation and current threats to European honeybees. Apidologie 2009, 40, 263–284. [Google Scholar] [CrossRef] [Green Version]
- Ilyasov, R.A.; Lee, M.-L.; Takahashi, J.-I.; Kwon, H.W.; Nikolenko, A.G. A revision of subspecies structure of western honey bee Apis mellifera. Saudi J. Biol. Sci. 2020, 27, 3615–3621. [Google Scholar] [CrossRef]
- Bouga, M.; Harizanis, P.C.; Kilias, G.; Alahiotis, S. Genetic divergence and phylogenetic relationships of honey bee Apis mellifera (Hymenoptera:Apidae) populations from Greece and Cyprus using PCR-RFLP analysis of three mtDNA segments. Apidologie 2005, 36, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, E.; Bouga, M.; Staykova, T.; Mladenovic, M.; Rasic, S.; Charistos, L.; Hatjina, F.; Petrov, P. The genetic variability of honey bees from the Southern Balkan Peninsula, based on alloenzymic data. J. Apic. Res. 2012, 51, 329–335. [Google Scholar] [CrossRef]
- Munoz, I.; Alice Pinto, M.; De la Rua, P. Temporal changes in mitochondrial diversity highlights contrasting population events in Macaronesian honey bees. Apidologie 2013, 44, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Uzunov, A.; Meixner, M.D.; Kiprijanovska, H.; Andonov, S.; Gregorc, A.; Ivanova, E.; Bouga, M.; Dobi, P.; Buechler, R.; Francis, R.; et al. Genetic structure of Apis mellifera macedonica in the Balkan Peninsula based on microsatellite DNA polymorphism. J. Apic. Res. 2014, 53, 288–295. [Google Scholar] [CrossRef]
- Ilyasov, R.A.; Poskryakov, A.V.; Petukhov, A.V.; Nikolenko, A.G. Molecular genetic analysis of five extant reserves of black honeybee Apis melifera melifera in the Urals and the Volga region. Russ. J. Genet. 2016, 52, 828–839. [Google Scholar] [CrossRef]
- Franck, P.; Garnery, L.; Celebrano, G.; Solignac, M.; Cornuet, J.M. Hybrid origins of honeybees from Italy (Apis mellifera ligustica) and Sicily (A. m. sicula). Mol. Ecol. 2000, 9, 907–921. [Google Scholar] [CrossRef]
- Bouga, M.; Alaux, C.; Bienkowska, M.; Buechler, R.; Carreck, N.L.; Cauia, E.; Chlebo, R.; Dahle, B.; Dall’Olio, R.; De la Rua, P.; et al. A review of methods for discrimination of honey bee populations as applied to European beekeeping. J. Apic. Res. 2011, 50, 51–84. [Google Scholar] [CrossRef] [Green Version]
- Canovas, F.; de la Rua, P.; Serrano, J.; Galian, J. Microsatellite variability reveals beekeeping influences on Iberian honeybee populations. Apidologie 2011, 42, 235–251. [Google Scholar] [CrossRef] [Green Version]
- Alice Pinto, M.; Henriques, D.; Chavez-Galarza, J.; Kryger, P.; Garnery, L.; van der Zee, R.; Dahle, B.; Soland-Reckeweg, G.; de la Rua, P.; Dall’Olio, R.; et al. Genetic integrity of the Dark European honey bee (Apis mellifera mellifera) from protected populations: A genome-wide assessment using SNPs and mtDNA sequence data. J. Apic. Res. 2014, 53, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Garnery, L.; Franck, P.; Baudry, E.; Vautrin, D.; Cornuet, J.-M.; Solignac, M. Genetic diversity of the west European honey bee (Apis mellifera mellifera and A. m. iberica). II. Microsatellite loci. Genet. Sel. Evol. 1998, 30, S49–S74. [Google Scholar] [CrossRef]
- Garnery, L.; Franck, P.; Baudry, E.; Vautrin, D.; Cornuet, J.-M.; Solignac, M. Genetic diversity of the west European honey bee (Apis mellifera mellifera and A. m. iberica). I. Mitochondrial DNA. Genet. Sel. Evol. 1998, 30, S31–S47. [Google Scholar] [CrossRef]
- Miguel, I.; Iriondo, M.; Garnery, L.; Sheppard, W.S.; Estonba, A. Gene flow within the M evolutionary lineage of Apis mellifera: Role of the Pyrenees, isolation by distance and post-glacial re-colonization routes in the western Europe. Apidologie 2007, 38, 141–155. [Google Scholar] [CrossRef]
- Meixner, M.D.; Worobik, M.; Wilde, J.; Fuchs, S.; Koeniger, N. Apis mellifera mellifera in eastern Europe-morphometric variation and determination of its range limits. Apidologie 2007, 38, 191–197. [Google Scholar] [CrossRef]
- Kandemir, I.; Ozkan, A.; Fuchs, S. Reevaluation of honeybee (Apis mellifera) microtaxonomy: A geometric morphometric approach. Apidologie 2011, 42, 618–627. [Google Scholar] [CrossRef] [Green Version]
- Parejo, M.; Wragg, D.; Gauthier, L.; Vignal, A.; Neumann, P.; Neuditschko, M. Using Whole-Genome Sequence Information to Foster Conservation Efforts for the European Dark Honey Bee, Apis mellifera mellifera. Front. Ecol. Evol. 2016, 4, 140. [Google Scholar] [CrossRef] [Green Version]
- Engel, M.S. The taxonomy of recent and fossil honey bees (Hymenoptera: Apidae; Apis). J. Hymenopt. Res. 1999, 8, 165–196. [Google Scholar]
- Foti, N. Researches on morphological characteristics and biological features of the bee population in Romania. In Proceedings of the XXth Jubiliar International Congress of Beekeeping Apimondia, Bucharest, Romania, 15–18 February 1965; pp. 171–176. [Google Scholar]
- Petrov, P. Systematics of Bulgarian Bees; Pchelarstvo: Sofia, Bulgaria, 1991. [Google Scholar]
- Ilyasov, R.; Nikolenko, A.; Tuktarov, V.; Goto, K.; Takahashi, J.-I.; Kwon, H.W. Comparative analysis of mitochondrial genomes of the honey bee subspecies A. m. caucasica and A. m. carpathica and refinement of their evolutionary lineages. J. Apic. Res. 2019, 58, 567–579. [Google Scholar] [CrossRef]
- Hassett, J.; Browne, K.A.; McCormack, G.P.; Moore, E.; Soland, G.; Geary, M.; Native Irish Honey Bee, S. A significant pure population of the dark European honey bee (Apis mellifera mellifera) remains in Ireland. J. Apic. Res. 2018, 57, 337–350. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M.; Kryger, P.; Meixner, M.; Bouga, M.; Ivanova, E.; Andonov, S.; Berg, S.; Bienkowska, M.; Büchler, R.; Charistos, L.; et al. The genetic origin of honey bee colonies used in the COLOSS Genotype-Environment Interactions Experiment: A comparison of methods. J. Apic. Res. 2014, 53, 188–204. [Google Scholar] [CrossRef]
- Evans, J.D.; Schwarz, R.S.; Chen, Y.P.; Budge, G.; Cornman, R.S.; De la Rua, P.; de Miranda, J.R.; Foret, S.; Foster, L.; Gauthier, L.; et al. Standard methods for molecular research in Apis mellifera. J. Apic. Res. 2013, 52, 1–54. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J. Isolation of Plant DNA from Fresh Tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Elsik, C.G.; Worley, K.C.; Bennett, A.K.; Beye, M.; Camara, F.; Childers, C.P.; de Graaf, D.C.; Debyser, G.; Deng, J.; Devreese, B.; et al. Finding the missing honey bee genes: Lessons learned from a genome upgrade. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data, P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Kofler, R.; Pandey, R.V.; Schloetterer, C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 2011, 27, 3435–3436. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Larranaga, O.; Langa, J.; Rendo, F.; Manzano, C.; Iriondo, M.; Estonba, A. Genomic selection signatures in sheep from the Western Pyrenees. Genet. Sel. Evol. 2018, 50, 9. [Google Scholar] [CrossRef] [Green Version]
- Gruening, B.; Dale, R.; Sjoedin, A.; Chapman, B.A.; Rowe, J.; Tomkins-Tinch, C.H.; Valieris, R.; Koester, J.; Team, B. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 2018, 15, 475–476. [Google Scholar] [CrossRef]
- Koester, J.; Rahmann, S. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Pearson, K. Note on Regression and Inheritance in the Case of Two Parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 3 February 2021).
- Kassambara, A. ggpubr: ‘ggplot2′ Based Publication Ready Plots. R Package Version 0.4.0. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 3 February 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evol. Int. J. Org. Evol. 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [Green Version]
- Meisner, J.; Albrechtsen, A. Inferring Population Structure and Admixture Proportions in Low-Depth NGS Data. Genetics 2018, 210, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Vieira, F.G.; Lassalle, F.; Korneliussen, T.S.; Fumagalli, M. Improving the estimation of genetic distances from Next-Generation Sequencing data. Biol. J. Linn. Soc. 2016, 117, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Skotte, L.; Korneliussen, T.S.; Albrechtsen, A. Estimating Individual Admixture Proportions from Next Generation Sequencing Data. Genetics 2013, 195, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Franck, P.; Garnery, L.; Solignac, M.; Cornuet, J.M. Molecular confirmation of a fourth lineage in honeybees from the Near East. Apidologie 2000, 31, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Roesti, M.; Salzburger, W.; Berner, D. Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evol. Biol. 2012, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Cridland, J.M.; Tsutsui, N.D.; Ramirez, S.R. The Complex Demographic History and Evolutionary Origin of the Western Honey Bee, Apis Mellifera. Genome Biol. Evol. 2017, 9, 457–472. [Google Scholar] [CrossRef] [Green Version]
- Fournier, T.; Abou Saada, O.; Hou, J.; Peter, J.; Caudal, E.; Schacherer, J. Extensive impact of low-frequency variants on the phenotypic landscape at population-scale. Elife 2019, 8, e49258. [Google Scholar] [CrossRef]
- Momozawa, Y.; Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 2021, 66, 11–23. [Google Scholar] [CrossRef]
- Anderson, E.C.; Skaug, H.J.; Barshis, D.J. Next-generation sequencing for molecular ecology: A caveat regarding pooled samples. Mol. Ecol. 2014, 23, 502–512. [Google Scholar] [CrossRef]
- Rode, N.O.; Holtz, Y.; Loridon, K.; Santoni, S.; Ronfort, J.; Gay, L. How to optimize the precision of allele and haplotype frequency estimates using pooled-sequencing data. Mol. Ecol. Resour. 2018, 18, 194–203. [Google Scholar] [CrossRef]
- Garnier-Géré, P.; Chikhi, L. Population Subdivision, Hardy-Weinberg Equilibrium and the Wahlund Effect. eLS 2013. [Google Scholar] [CrossRef]
- Kristensen, T.N.; Pedersen, K.S.; Vermeulen, C.J.; Loeschcke, V. Research on inbreeding in the ‘omic’ era. Trends Ecol. Evol. 2010, 25, 44–52. [Google Scholar] [CrossRef]
- Parejo, M.; Wragg, D.; Henriques, D.; Charriere, J.-D.; Estonba, A. Digging into the Genomic Past of Swiss Honey Bees by Whole-Genome Sequencing Museum Specimens. Genome Biol. Evol. 2020, 12, 2535–2551. [Google Scholar] [CrossRef]
- Parejo, M.; Wragg, D.; Henriques, D.; Vignal, A.; Neuditschko, M. Genome-wide scans between two honeybee populations reveal putative signatures of human-mediated selection. Anim. Genet. 2017, 48, 704–707. [Google Scholar] [CrossRef] [Green Version]
- Henriques, D.; Wallberg, A.; Chavez-Galarza, J.; Johnston, J.S.; Webster, M.T.; Alice Pinto, M. Whole genome SNP-associated signatures of local adaptation in honeybees of the Iberian Peninsula. Sci. Rep. 2018, 8, 11145. [Google Scholar] [CrossRef] [Green Version]
- Wragg, D.; Marti-Marimon, M.; Basso, B.; Bidanel, J.-P.; Labarthe, E.; Bouchez, O.; Le Conte, Y.; Vignal, A. Whole-genome resequencing of honeybee drones to detect genomic selection in a population managed for royal jelly. Sci. Rep. 2016, 6, 27168. [Google Scholar] [CrossRef] [Green Version]
- Bastide, H.; Betancourt, A.; Nolte, V.; Tobler, R.; Stoebe, P.; Futschik, A.; Schloetterer, C. A Genome-Wide, Fine-Scale Map of Natural Pigmentation Variation in Drosophila melanogaster. PLoS Genet. 2013, 9, e1003534. [Google Scholar] [CrossRef] [Green Version]
- Beissinger, T.M.; Hirsch, C.N.; Vaillancourt, B.; Deshpande, S.; Barry, K.; Buell, C.R.; Kaeppler, S.M.; Gianola, D.; de Leon, N. A Genome-Wide Scan for Evidence of Selection in a Maize Population Under Long-Term Artificial Selection for Ear Number. Genetics 2014, 196, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Lattorff, H.M.G.; Buchholz, J.; Fries, I.; Moritz, R.F.A. A selective sweep in a Varroa destructor resistant honeybee (Apis mellifera) population. Infect. Genet. Evol. 2015, 31, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, C.W.; Behura, S.K.; Berlocher, S.H.; Clark, A.G.; Johnston, J.S.; Sheppard, W.S.; Smith, D.R.; Suarez, A.V.; Weaver, D.; Tsutsui, N.D. Thrice out of Africa: Ancient and recent expansions of the honey bee, Apis mellifera. Science 2006, 314, 642–645. [Google Scholar] [CrossRef] [Green Version]
- Puskadija, Z.; Kovacic, M.; Raguz, N.; Lukic, B.; Presern, J.; Tofilski, A. Morphological diversity of Carniolan honey bee (Apis mellifera carnica) in Croatia and Slovenia. J. Apic. Res. 2020, 60, 326–336. [Google Scholar] [CrossRef]
- Harpur, B.A.; Minaei, S.; Kent, C.F.; Zayed, A. Management increases genetic diversity of honey bees via admixture. Mol. Ecol. 2012, 21, 4414–4421. [Google Scholar] [CrossRef]
- Chavez-Galarza, J.; Henriques, D.; Johnston, J.S.; Carneiro, M.; Rufino, J.; Patton, J.C.; Alice Pinto, M. Revisiting the Iberian honey bee (Apis mellifera iberiensis) contact zone: Maternal and genome-wide nuclear variations provide support for secondary contact from historical refugia. Mol. Ecol. 2015, 24, 2973–2992. [Google Scholar] [CrossRef]
- Miguel, I.; Baylac, M.; Iriondo, M.; Manzano, C.; Garnery, L.; Estonba, A. Both geometric morphometric and microsatellite data consistently support the differentiation of the Apis mellifera M evolutionary branch. Apidologie 2011, 42, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Browne, K.A.; Hassett, J.; Geary, M.; Moore, E.; Henriques, D.; Soland-Reckeweg, G.; Ferrari, R.; Mac Loughlin, E.; O’Brien, E.; O’Driscoll, S.; et al. Investigation of free-living honey bee colonies in Ireland. J. Apic. Res. 2020, 60, 229–240. [Google Scholar] [CrossRef]
- Nielsen, E.S.; Henriques, R.; Toonen, R.J.; Knapp, I.S.S.; Guo, B.; von der Heyden, S. Complex signatures of genomic variation of two non-model marine species in a homogeneous environment. BMC Genom. 2018, 19, 347. [Google Scholar] [CrossRef]
- Kurland, S.; Wheat, C.W.; Mancera, M.d.l.P.C.; Kutschera, V.E.; Hill, J.; Andersson, A.; Rubin, C.-J.; Andersson, L.; Ryman, N.; Laikre, L. Exploring a Pool-seq-only approach for gaining population genomic insights in nonmodel species. Ecol. Evol. 2019, 9, 11448–11463. [Google Scholar] [CrossRef]
- Canovas, F.; De la Rua, P.; Serrano, J.; Galian, J. Analysis of a contact area between two distinct evolutionary honeybee units: An ecological perspective. J. Insect Conserv. 2014, 18, 927–937. [Google Scholar] [CrossRef]
- Nazzi, F. Morphometric analysis of honey bees from an area of racial hybridization in northeastern Italy. Apidologie 1992, 23, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Cornuet, J.-M.; Excoffier, L.; Franck, P.; Estoup, A. Bayesian Inference under Complex Evolutionary Scenarios Using Microsatellite Markers: Multiple Divergence and Genetic Admixture Events in the Honey Bee, Apis mellifera. Genetic Diversity; Mahoney, C.L., Springer, D.A., Eds.; Nova Science Publishers: New York, NY, USA, 2009. [Google Scholar]
- Janczyk, A.; Meixner, M.D.; Tofilski, A. Morphometric identification of the endemic Maltese honey bee (Apis mellifera ruttneri). J. Apic. Res. 2021, 60, 157–164. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Z.; Pan, Q.; Chen, X.; Wang, H.; Guo, H.; Liu, S.; Lu, H.; Tian, S.; Li, R.; et al. Genomic Analyses Reveal Demographic History and Temperate Adaptation of the Newly Discovered Honey Bee Subspecies Apis mellifera sinisxinyuan n. ssp. Mol. Biol. Evol. 2016, 33, 1337–1348. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, H.; Liu, Z.; Chen, X.; Tang, J.; Meng, F.; Shi, W. Population Genomics Provide Insights into the Evolution and Adaptation of the Eastern Honey Bee (Apis cerana). Mol. Biol. Evol. 2018, 35, 2260–2271. [Google Scholar] [CrossRef]
- Steinrucken, M.; Kamm, J.; Spence, J.P.; Song, Y.S. Inference of complex population histories using whole-genome sequences from multiple populations. Proc. Natl. Acad. Sci. USA 2019, 116, 17115–17120. [Google Scholar] [CrossRef] [Green Version]
- Steiner, C.C.; Putnam, A.S.; Hoeck, P.E.A.; Ryder, O.A. Conservation Genomics of Threatened Animal Species. Annu. Rev. Anim. Biosci. 2013, 1, 261–281. [Google Scholar] [CrossRef] [Green Version]
- Fleming, D.S.; Koltes, J.E.; Fritz-Waters, E.R.; Rothschild, M.F.; Schmidt, C.J.; Ashwell, C.M.; Persia, M.E.; Reecy, J.M.; Lamont, S.J. Single nucleotide variant discovery of highly inbred Leghorn and Fayoumi chicken breeds using pooled whole genome resequencing data reveals insights into phenotype differences. BMC Genom. 2016, 17, 812. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | Population | Subspecies | Country | Pool Sequencing Samples (N) | Individual Sequencing Samples (N) | Origin of Samples/References |
|---|---|---|---|---|---|---|
| M | ibe_esp_eus | A. m. iberiensis | Spain | 100 | 10 | Miguel et al., 2007 [35] |
| mel_irl | A. m. mellifera | Ireland | 100 | 10 | Hassett et al., 2018 [43] | |
| mel_rus | A. m. mellifera | Russia (Ural) | 100 | 10 | This study, Momeni et al., 2021 [17] | |
| C | car_aut_hun | A. m. carnica | Austria & Hungary | 100 | 10 | This study, Momeni et al., 2021 [17] |
| rod_bgr | A. m. rodopica | Bulgaria | 95 | 10 | This study, Momeni et al., 2021 [17] | |
| carp_rou_mda | A. m. carpatica | Romania & Moldova | 90 | 10 | This study, Momeni et al., 2021 [17] | |
| mac_mkd_grc | A. m. macedonica | North Macedonia & N-Greece | 86 | 10 | This study, Uzunov et al., 2014 [27] | |
| cec_grc | A. m. cecropia | Greece | 93 | 10 | This study, Momeni et al., 2021 [17] | |
| ada_grc | A. m. adami | Greece (Crete) | 88 | 9 | This study, Momeni et al., 2021 [17] | |
| O | cyp_cyp | A. m. cypria | Cyprus | 100 | 10 | This study, Momeni et al., 2021 [17] |
| ana_tur | A. m. anatoliaca | Turkey | 100 | 10 | This study, Francis et al., 2014 [44] | |
| rem_arm | A. m. remipes | Armenia | 90 | 10 | This study, Momeni et al., 2021 [17] | |
| cau_tur_geo | A. m. caucasia | NE-Turkey & Georgia | 105 | 10 | This study, Momeni et al., 2021 [17] | |
| A | rut_mlt | A. m. ruttneri | Malta | 100 | 10 | This study, Momeni et al., 2021 [17] |
| TOTAL | 1347 | 139 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Parejo, M.; Momeni, J.; Langa, J.; Nielsen, R.O.; Shi, W.; SMARTBEES WP3 DIVERSITY CONTRIBUTORS; Vingborg, R.; Kryger, P.; Bouga, M.; et al. Population Structure and Diversity in European Honey Bees (Apis mellifera L.)—An Empirical Comparison of Pool and Individual Whole-Genome Sequencing. Genes 2022, 13, 182. https://doi.org/10.3390/genes13020182
Chen C, Parejo M, Momeni J, Langa J, Nielsen RO, Shi W, SMARTBEES WP3 DIVERSITY CONTRIBUTORS, Vingborg R, Kryger P, Bouga M, et al. Population Structure and Diversity in European Honey Bees (Apis mellifera L.)—An Empirical Comparison of Pool and Individual Whole-Genome Sequencing. Genes. 2022; 13(2):182. https://doi.org/10.3390/genes13020182
Chicago/Turabian StyleChen, Chao, Melanie Parejo, Jamal Momeni, Jorge Langa, Rasmus O. Nielsen, Wei Shi, SMARTBEES WP3 DIVERSITY CONTRIBUTORS, Rikke Vingborg, Per Kryger, Maria Bouga, and et al. 2022. "Population Structure and Diversity in European Honey Bees (Apis mellifera L.)—An Empirical Comparison of Pool and Individual Whole-Genome Sequencing" Genes 13, no. 2: 182. https://doi.org/10.3390/genes13020182